Gaussian09W破解版[安装教程]_Gaussian09W v9.5破解版下载
Gaussian 09W是一款全球知名的软件应用,在行业内享有极高的知名度,相信很多业内人士对它并不陌生,该软件拥有非常干净整洁的软件界面和排版布局合理明了的功能,可以轻松的帮助用户上手并且使用这款软件,软件的功能很齐全,出彩的设计,优质的体验,无一不再说这款软件的强大之处,知识兔小编为大家带来Gaussian 09下载,并且可以为使用这款软件的用户分享相关教程以及安装方法,希望可以帮到各位需要此软件的网友。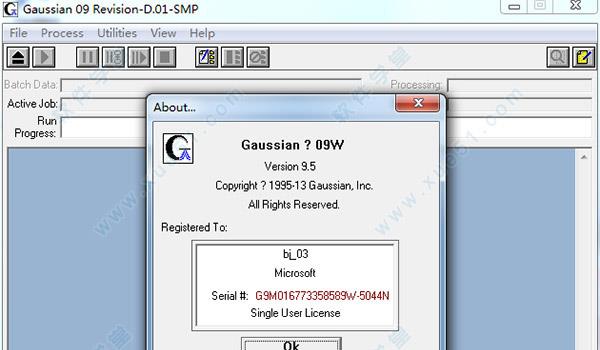
Gaussian 09W是一个功能强大的量子化学综合软件包,可以进行振动频率分析,以便预测红外/拉曼光谱和正常模式,还分享了各种其他光谱,包括振动光谱。Gaussian 09W被用作帮助探索分子系统和化学反应的工具,具有过渡态能量和结构、键和反应能量、分子轨道、原子电荷和电势、振动频率、红外和拉曼光谱、核磁性质、极化率和超极化率、热力学性质、反应路径,计算可以对体系的基态或激发态执行,并可以预测周期体系的能量,结构和分子轨道,可用于研究许多化学领域的课题,例如取代基的影响,化学反应机理,势能曲面和激发能等等。同时软件可执行程序可在不同型号的大型计算机,超级计算机,工作站和个人计算机上运行,并相应有不同的版本。知识兔小编给大家分享的是Gaussian 09W破解版,数据包内附带注册码序列号,通过使用它可以完美进行激活破解软件,解锁软件中被限制的很多功能,用户就可以免费、无功能限制使用了。本文有图文结合详细的安装教程(附破解教程)可供大家参考,大家可以参照具体教程步骤进行软件安装、破解操作,希望对大家有帮助,有需求或者喜欢这款软件的用户欢迎前来3D软件下载体验。
安装教程(附破解教程)
1.下载并解压安装包压缩包,然后知识兔来进行安装教程(附破解教程)操作,双击运行“Setup.exe”程序进行软件安装。
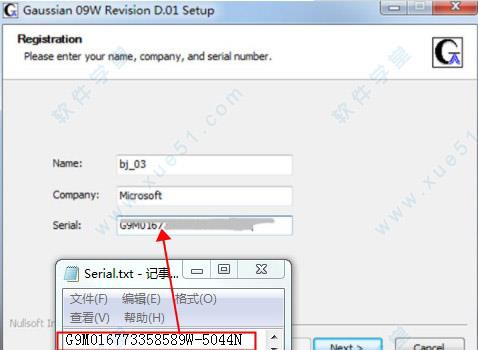
2.填写用户信息和注册码,用户信息可随意填写,注册码序列号Serial处填写软件安装包附带的Serial.txt记事本内注册码。
3.验证序列号,点击“确定”即可。
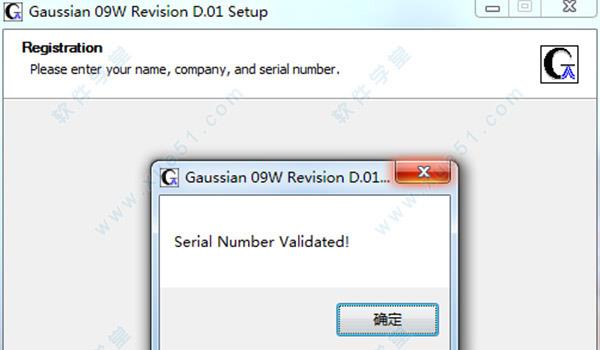
4.安装组件按照默认设置即可。
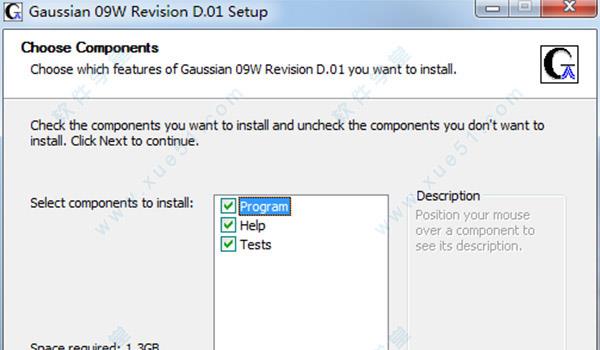
5.选择软件安装目录,可点击“Browse”更改,也可按照默认安装目录。
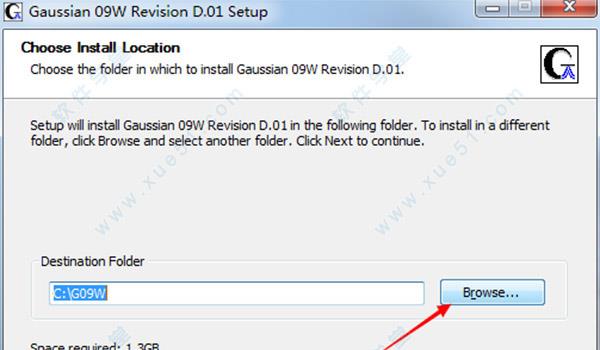
6.点击“install”开始安装软件。
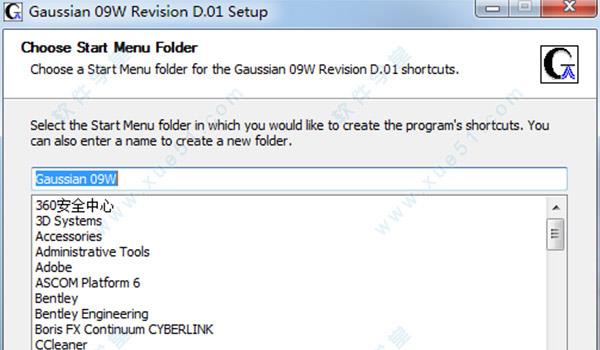
7.软件进入安装状态,安装过程需要一些时间,请大家耐心稍待片刻。
8.软件安装完成,点击“finish”退出安装程序。
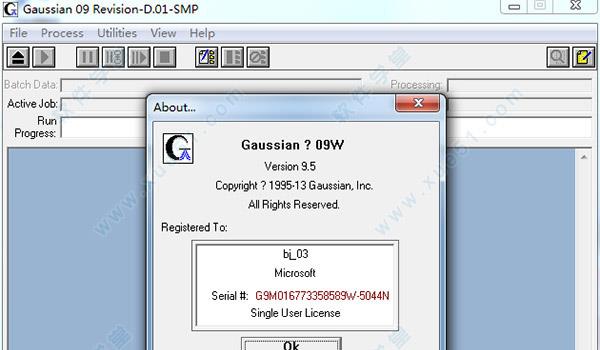
9.运行软件,软件提示已授权,表明激活成功。

10.进入软件主界面,查看软件信息,知识兔可以看到软件状态已显示激活,至此,软件成功破解,用户可以无限制、免费使用啦!
版本功能
能量和求导
⑴、最近发展的半经验模型(AM1,PM3,PM3MM,PDDG,PM6),计算解析一阶导和二阶导,用户自定义参数,以及结合使用PCM溶剂模型。
⑵、TDDFT解析梯度和数值频率。
⑶、EOM-CCSD计算激发能。
⑷、新的DFT泛函,包括HSE,wB97,m05/m06,LC类泛函,以及双杂化B2PLYP。
⑸、经验离散模型和相应的泛函。
⑹、ROMP3,ROMP4,ROCCSD,ROCCSD(T)能量。
⑺、W1RO,W1BD,G4方法计算能量。
⑻、DFTB半经验模型,以及使用解析矩阵元的DFTBA版本。
ONIOM
⑴、ONIOM与PCM组合。有多种ONIOM+PCM模型。
⑵、ONIOM计算IRC,即使分子包含上千个原子效率也很高。
溶剂化
⑴、新的PCM溶剂化算法,使能量成为核坐标的连续函数。现在,PCM的几何优化与气相优化的收敛速度一样。
⑵、特定态的自洽溶剂化,用于模拟荧光和其它发射过程。它对上百种溶剂进行了参数化,可以给出非常好的总溶剂化自由能。
几何优化和IRC
⑴、能量最小化默认使用GEDⅡS几何优化算法,这对大的柔软分子特别有帮助。
⑵、对最小值和过渡结构使用二次收敛ONIOM(MO:MM)优化,既用于力学部分,也用于电子嵌入部分。
⑶、一个输入部分,用于控制优化中的冻结或非冻结原子。可以用原子、元素、残基、或ONIOM层来指定原子。
分子特性
⑴、解析的含频ROA强度。
⑵、解析的DFT 超极化率。
⑶、使用两个态的谐振模式,通过Franck-Condon原理计算电子激发、发射、光电离的谱带带型。
⑷、用Herzberg-Teller或Franck-Condon-Herzberg-Teller理论计算电子激发的谱带带型。
⑸、选择简正模式用于显示,非谐校正,和FC/HT/FCHT分析。可通过原子、元素、残基、或ONIOM层来选择。
分析和输出
⑴、蛋白质二级结构的信息可以包含在分子指定输入部分,或者.fchk文件中。
⑵、轨道布居分析,给出原子或角动量对轨道的贡献。
⑶、正则UHF/UDFT进行二次正交化,用于显示或用于ROHF计算的初始猜测。
⑷、CIS和TD激发的自然跃迁轨道分析。
⑸、把占据轨道投影到最小基之后,进行Mulliken布居分析。当基组增大时,这能给出稳定的布居。
其它新增功能
⑴、SCF的初始猜测可以从片段计算的组合产生,需要指定每个片段的电荷和自旋。
⑵、用四点差分而不是默认的两点差分计算数值频率,具有更高的精度和数值稳定性。
效率改善
⑴、HF和DFT对大分子的频率计算更快,特别是当并行时。
⑵、FMM以及线性标度的库仑和交换对簇并行。
⑶、大体系的ONIOM(MO:MM)频率计算更快,特别是对电子嵌入。可以计算100-200 QM原子和6000 MM原子的频率。
⑷、在大型频率计算中保存简正模式,用于显示或打印模式,以及开始IRC=RCFC任务。
⑸、CC,BD,和EOM-CCSD振幅可以保存在检查点文件中,在以后的计算中读入。保存BD轨道并在以后读入。
⑹、半经验,HF,和DFT的频率计算可以在中期计算中重新开始。
⑺、CC和EOM-CC计算可以在中期计算中重新开始。
⑻、ONIOM各步的初始猜测可以来自不同的检查点文件。
⑼、加入了SVP,TZVP,QZV基组的密度拟合基。Fit关键字调用与AO基组匹配的拟合基,没有特定的拟合基时需要Auto关键字。
⑽、在Default.Route文件中包含DensityFit关键字,只要执行纯密度泛函就使用拟合。
⑾、为了与文献发表的基组兼容,读入的密度拟合因子可以是非归一化的原函数,密度归一化的原函数,或归一化的原函数。
特色亮点
1.振动频率和正常模式, 包括显示输出限制指定原子/残留/模式(可选模式排序)。
2.可重新开始的分析高频和DFT频率。
3.非谐频率分析包括 完全不和谐的红外intensitie,DCPT2 HDCPT2 resonance-free方法计算非谐频率和配分函数,和电子振动的计算包括儿童早期开发。
4.莫:MM ONIOM频率包括电子嵌入。
5.分析红外和 静态和动态拉曼强度(高频& DFT;MP2 IR)。
6.Pre-resonance拉曼光谱(高频和DFT)。
7.预计频率垂直于反应路径。
8.核磁共振屏蔽张量& GIAO磁脆弱的感情(高频,DFT,MP2)和 增强自旋自旋耦合(高频,DFT)。
9.专业基础集核磁共振自旋自旋耦合计算。
10.振动圆二色性(VCD)旋转的优势(高频和DFT)
11.动态拉曼旋光性(ROA)强度。
12.拉曼和ROA计算可以分开进行频率分析与更大的基础设置(如推荐的文献)。
13.谐波振动转动耦合。
14.增强不和谐的振动分析。
15.不和谐的振动转动耦合通过微扰理论。
16.阻碍了转子分析。
下载仅供下载体验和测试学习,不得商用和正当使用。

![PICS3D 2020破解版[免加密]_Crosslight PICS3D 2020(含破解补丁)](/d/p156/2-220420222641552.jpg)


![AutoCAD LT 2023破解版[免加密]_AutoCAD LT 2023.1 授权激活版](/d/p156/2-22033009222N55.jpg)