DS BIOVIA Materials Studio 2023 v23.1.0.3829 x64 化学材料科学建模和模拟工具下载
BIOVIA Materials Studio 2023 是 BIOVIA 公司最新版本的化学和材料科学建模和模拟科学工具。Materials Studio 能够帮助研究人员理解材料的分子或晶体结构与性质之间的关系。最新版本为科学家分享了广泛的世界级求解器和参数集,从量子到微观尺度运行。此版本包含了全范围求解器的增强功能,为粗粒化原子模型分享了更多便利,并显著提高了经典模拟的性能。BIOIVA Materials Studio 2023 让我们比以往更准确、更方便地模拟更多材料属性。
用于粗粒化模拟
自动粗粒化和珠型化
粗粒化分子动力学(CGMD),其中一组原子由单个珠子表示,越来越受欢迎,用于探测软物质系统从几十到几百纳米的结构和性质——这些尺度对于常规原子模拟来说是无法达到的。成功的 CGMD 模拟需要一组经过良好验证的力场参数来描述珠子之间的相互作用,并为每个珠子分配适当的力场类型。
在过去的 20 年中,Martini 力场[1-3]的方法已经证明非常受欢迎,并适用于广泛的材料,最近的 Martini 3 版本[4,5]可以声称分享迄今为止最准确的表示。Martini 3 力场可作为 Mesocite 模块的一部分使用,并且知识兔 Materials Studio 2023 分享了一组新功能,知识兔可以自动将结构从原子模型粗粒化。这大大简化了分配 Martini 3 分享的珠型所需的复杂步骤。这具有建模效率和减少珠型错误的双重好处。这些工具既可以从新脚本菜单访问,知识兔也可以以 Pipeline Pilot 协议的形式在 Materials Studio Collection 中访问。协议的包含使配置定制粗粒化过程非常高效。
新工具!
Martini 3 工具经过改进,现在具有新的 MS Martini 3 粗粒化器功能。它直接从原子模型(见图1)生成中观结构模型,包括珠映射和力场类型,只需使用一个模板研究表。知识兔可以通过 Materials Studio Visualizer 指定自定义交互的模板文件,例如图2中所示的那些。这些模板允许复杂情况(如涉及环的铰接结构)得到表示(其中珠心通常不位于原子中心)。
分享了一个定制的 MS Martini 3 力场,包括所有必需的价项和中观结构模型,准备好用 Mesocite 进行模拟。当与 NVIDIA GPU 一起使用时,Mesocite 粗粒分子动力学也能获得显著加速。
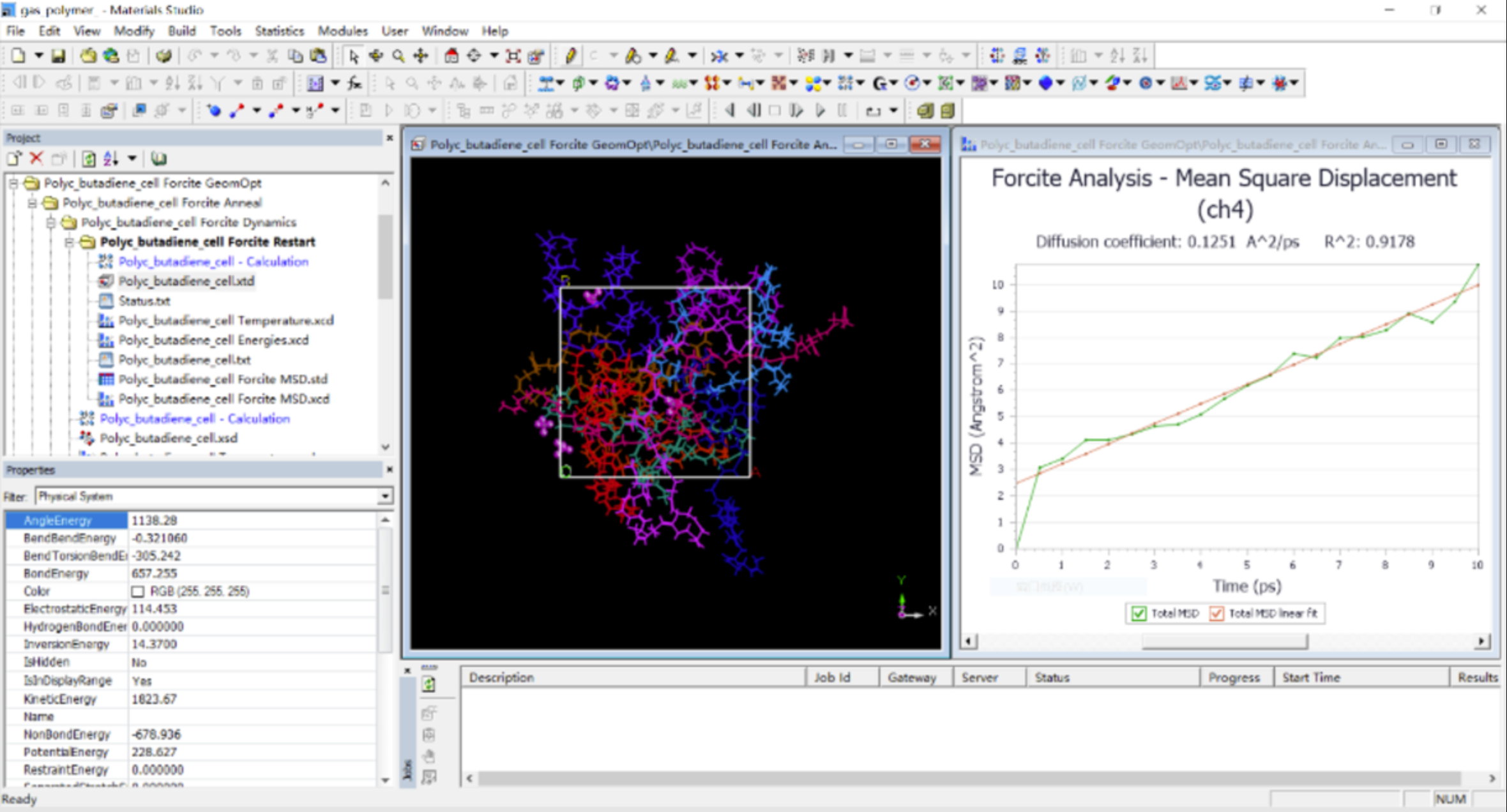
介观耗散粒子动力学
Mesocite 耗散粒子动力学(DPD)是一种流行且经过验证的方法,用于研究聚合物混合物、表面活性剂系统等软凝聚态物质的自组装过程。DPD 中体现的软势方法分享了一种非常有效的方法来达到系统的热力学平衡状态。这些材料的重要子类包括聚电解质和带电表面活性剂系统,通过在 Mesocite DPD 中包含电荷珠子之间的静电相互作用,Materials Studio 2023 可以更准确地表示这些材料。
Mesocite DPD 现在通过允许使用高斯分布的电荷来模糊珠子电荷来支持静电学。这避免了当 DPD 珠子(由软有限势表示)穿过彼此时,不同电荷塌缩到奇点。在 Materials Studio 2023 中,我们还可以通过在 NVIDIA GPU 上运行 Mesocite DPD 来获得更快的动力学,并增加了两个新的分析功能:
- 从密度场计算结构因子。
- 压力分析,分享各向异性分量。
在 Materials Studio 2023 中,Mesocite DPD 模块分享了更快速地模拟更多系统的新功能。
用于金属合金设计
BIOVIA Materials Studio 2023 是一种用于金属合金设计的工具。它基于 OpenPhase_Core 求解器,于 Materials Studio 2021 中引入,用于模拟硬材料微观结构。使用 PhaseField 模块,我们可以通过 Pipeline Pilot Connector 中易于使用的界面方便地定义组分相、晶粒设置、热力学和动力学输入以及温度和压力条件。这允许详细研究多组分金属合金中的相变。
能够准确预测相变是提高使用添加制造工艺创建的金属部件质量的关键。金属晶粒微观结构的大小和形状对液态金属在打印过程中固化的速率非常敏感。在固化过程中可能会出现不同比例的晶相,最终影响材料性能。Materials Studio 2023 分享了用于研究这些转变的新功能,包括:
新协议!
分享了一种新协议来计算连续冷却转换(CCT)图,可用于预测合金的临界冷却速率。该协议可以利用共同安装的 ThermoCalc 或 OpenCalphad 软件包分享 CALPHAD 热力学输入。
新功能!
时间-温度-转换(TTT)图协议现在拟合并返回每个模拟温度的 Johnson-Mehl-Avrami(JMA)系数。JMA 模型描述了固体在恒定温度下从一相转变为另一相的方式。
用于化学反应
Materials Studio 已经分享了广泛的工具和功能,通过应用量子力学求解器(DMol3、CASTEP、DFTB+ 和 VAMP)、应用 FlexTS 模块进行化学过渡态搜索以及反应动力学预测(Cantera 和 Kinetix 模块)来理解化学反应。Materials Studio 2023 为使用 Materials Studio 来理解化学反应的科学家分享了进一步的能力,包括访问 TURBOMOLE 和它分享超越DFT方法。
新协议!
现在分享了一组协议,通过 Pipeline Pilot 协议对话框分享 TURBOMOLE 工作流,分享 DFT、xTB 和波函数方法。
- 所有方法的单点能计算
- 几何优化(使用 xTB、DFT 或 MP2)
- 过渡态优化(使用 xTB 或 DFT)
- IR 光谱/振动频率(使用 DFT)
- 极化率(使用 DFT)
- 光谱(使用 DFT)
新实用工具!
协议增强现在使构建自定义工作流以自动化反应模拟研究更加简单。
- 一个新的“Edit Sets”组件,允许我们在 Pipeline Pilot 中创建和管理集合。
- 脚本现在能够识别分子和片段的 SMILES 和化学名称。
- 一个用于分析模拟单元格内容或轨迹过程中不同试剂的发展情况的脚本可从新脚本菜单中获取
用于XPS光谱分析
XPS(X射线光电子能谱)测量分子或周期结构中核心电子的激发(结合)能量。Pipeline Pilot Materials Studio Collection 2023 中分享了一种新工具,知识兔可以方便地分享光谱。该协议还分享了通过计算或实验数据方便地分配偏移量的功能。
Materials Studio is a powerful molecular simulation software. Chemists, researchers and students of chemistry can use this program with a variety of molecular structures such as polymers (dendrimers, alloys, copolymers, homopolymers), nanostructures such as carbon nanotubes, nanomechanical equipment, types of compounds and inorganic crystals, organic structures and crystals. And … to simulate. Users can perform all of the mentioned structures in different molecular states to measure electronic structure, to measure static and dynamic structures. The program is capable of performing a variety of famous and important simulations, including quantum simulations including topics such as finding the optimal structure, finding transition states by DFT and Fock techniques.
At the molecular level, molecular dynamics and Monte Carlo dynamics can also be used for simulation. Other simulations such as DPD simulation, simulation of vapor-liquid and liquid-liquid gas equilibrium can also be performed. An important feature of this program is its modular structure. All the simulations are done by different modules. Modules consist of three parts: installation and configuration, implementation and analysis, which can be used to prepare the output file of the simulation, run the simulation and finally see the results of its analysis.
The program is graphically designed and combines all molecular structures and final analysis with 3D graphics to better understand the structures and analyzes presented. The program is capable of making useful predictions about the effectiveness of chemical actions and their behavior in chemical constituents. It has powerful statistical tools for displaying complex molecular relationships, and has a large database of different materials, making you completely unnecessary accessory. Using this software makes manual calculations and duplication work to a minimum, which in the long run reduces project costs and risk.
System Requirements Materials Studio
OS Windows: Microsoft Windows 11/Windows 10 Professional/Enterprise
软件下载地址:
DS BIOVIA Materials Studio 2023 v23.1.0.3829 x64 化学材料科学建模和模拟工具
Download 百度网盘:此内容仅限VIP查看,请先登录免责声明:根据我国《计算机软件保护条例》第十七条规定:“为了学习和研究软件内含的设计思想和原理,通过安装、显示、传输或者存储软件等方式使用软件的,知识兔可以不经软件著作权人许可,不向其支付报酬。”您需知晓知识兔所有内容资源均来源于网络,仅供用户交流学习与研究使用,版权归属原版权方所有,版权争议与知识兔无关,用户本人下载后不能用作商业或非法用途,需在24小时之内删除,否则后果均由用户承担责任。
下载仅供下载体验和测试学习,不得商用和正当使用。

![PICS3D 2020破解版[免加密]_Crosslight PICS3D 2020(含破解补丁)](/d/p156/2-220420222641552.jpg)